Pharmaceuticals & drug discovery
The pharmaceutical environment whether it is research and development (R&D) or quality assurance/control (QA/QC) in manufacturing put extreme demands on safety, quality and precision on any analysis tool. In R&D, the industry accepted drug discovery methods include single crystal diffraction and powder X-ray diffraction (pXRD). For tablet research and quality control in production, X-ray CT is a commonly used tool. Excillum X-ray sources help to further improve throughput, speed and image resolution beyond what has previously been possible both in manufacturing and laboratory environments.
To enhance sample throughput and speed of drug development, Excillum X-ray sources make it possible to perform on-site laboratory diffraction experiments faster than with conventional X-ray tubes. Therefore they can be used to screen and measure large sets of samples. Thanks to the small and focused beam capability of the X-ray source also small crystals and weakly diffracting crystals can be accurately analyzed. This leads to faster feedback and accelerated development of new drugs. In addition, due to the extremely small and bright X-ray spot, Excillum X-ray sources can be used to image multi-layer coating tablets, capsules and soft pills, offering unprecedented high-precision information on internal structure.

Application examples
Tin (IV) compunds
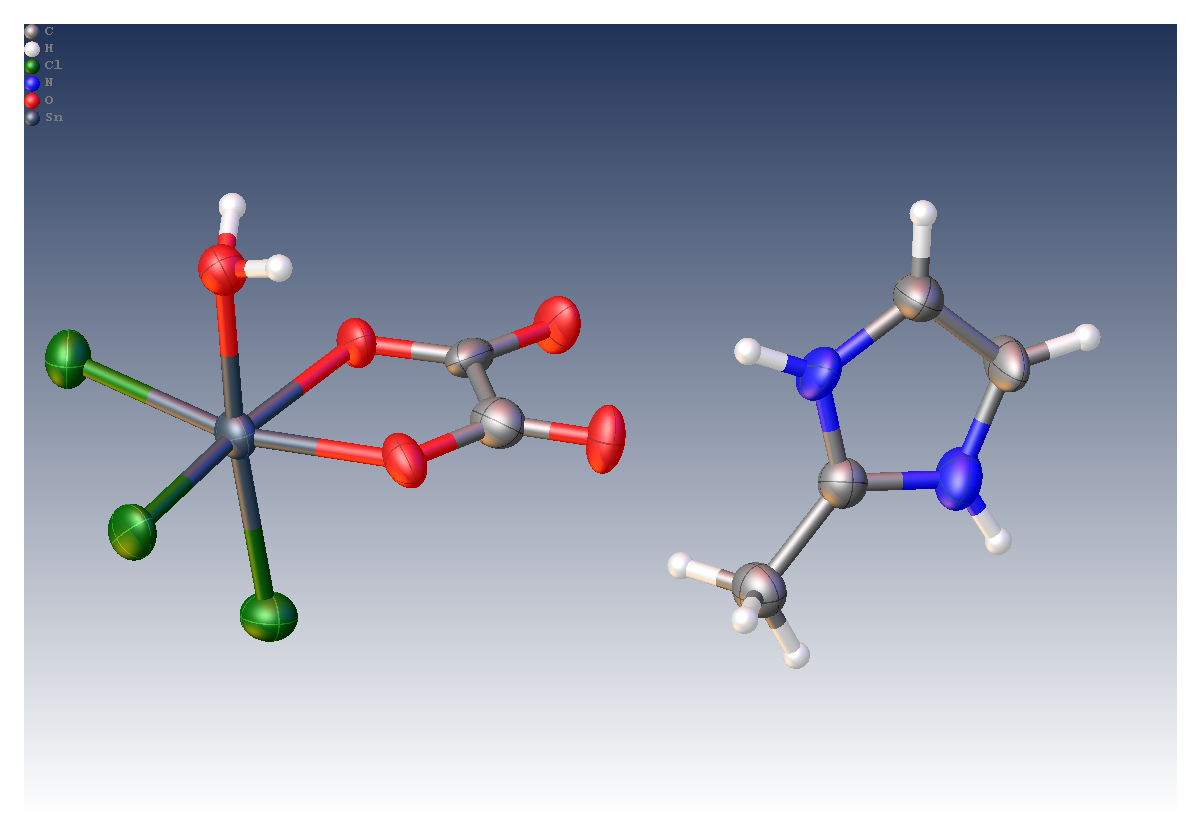
Tin (IV) compounds are interesting as potential catalysts and pharmaceuticals due to their biological activity. As part of a drive to understand these compounds, researchers at the Universities of Montreal, Cheikh Anta Diop and Bourgogne determined the crystal structure of a 50 μm crystal of [Sn(C2O4)Cl3(H2O)].(C4H7N2) using a MetalJet X-ray source.
- Crystal size: 0.05 x 0.04 x 0.04 mm3
- R1 = 6.2%
Absolute structures
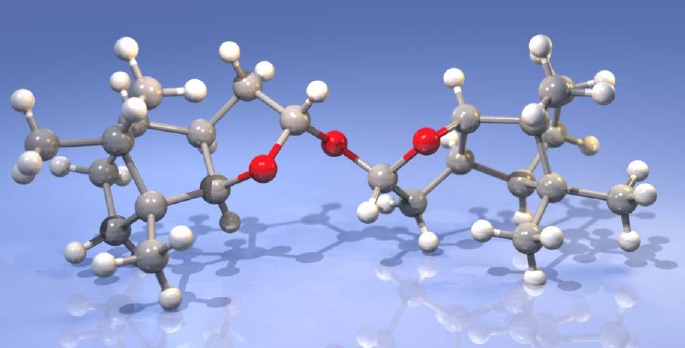
Application scientists at Bruker AXS used an Excillum MetalJet integrated into a Bruker D8 VENTURE diffractometer to successfully determine the absolute configuration of (2S)-(−)-2,2’-Oxybis(octahydro-7,8,8-trimethyl-4,7-methanobenzofuran), a light atom structure, where the heaviest atoms are three oxygen atoms.
- Flack x = 0.024(39) (Parsons‘)
- Crystal size: 0.15 x 0.05 x 0.04 mm3
- Experiment time: 4 hours
- Resolution: 0.75 Å
- Completeness: 96%
- Redundancy: 4
- R1 = 3.18%
Structural investigation of protein samples
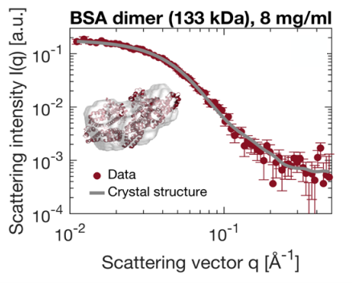
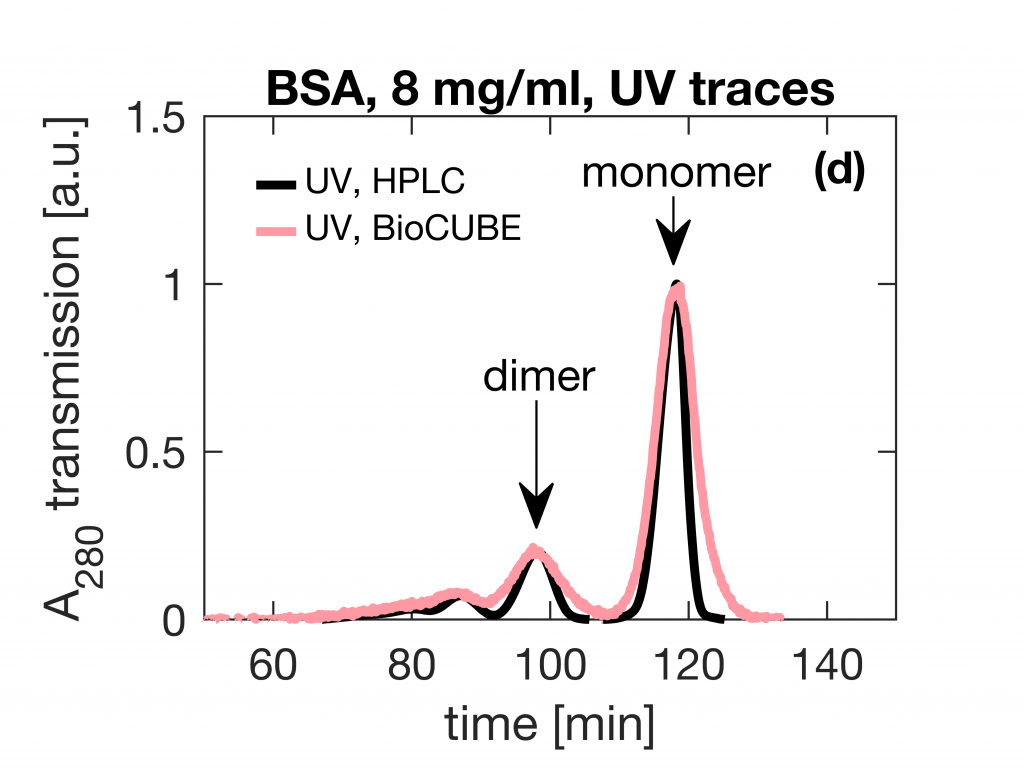
Small-Angle X-ray Scattering (SAXS) is a prevalent technique in structural biology for the investigation of protein structure and interactions. It gives efficient access to parameters like the radius of gyration, molecular weight, degree of folding, a low-resolution model of the 3D shape of proteins, protein multimers and complexes. Often, biological samples consist of a complex mixture of structurally different molecules and complexes in dynamic equilibria. SAXS measurements of such a mixture result in an average structure of the species in solution which can be very difficult – if not impossible – to deconvolute. An efficient solution to this problem is offered by the use of Size Exclusion Chromatography (SEC). By fractionating the mixture based on the size of the components, you can measure the SAXS signal of the individual species. The combination of SEC with SAXS does however put a huge time constraint on the SAXS measurements. Short measurement times are required to prevent the separated samples from reaching a new equilibrium. Short measurements on weakly scattering samples could previously only be done at the synchrotron. With the introduction of the MetalJet X-ray source, this measurement capability is now also available in the laboratory as was demonstrated with the Xenocs BioXolver. The images below present a case where Bovine Serum Albumin (BSA) was separated into the monomeric and dimeric form. The UV trace shows the separation into monomers and dimers. After separation, these species are measured with SAXS using an exposure time of only 60 seconds. As illustrated below, these short measurements provide sufficient statistics both for concentrations of 8 mg/ml as well as down to 1 mg/ml to establish the most important parameters and obtain a 3D structure.
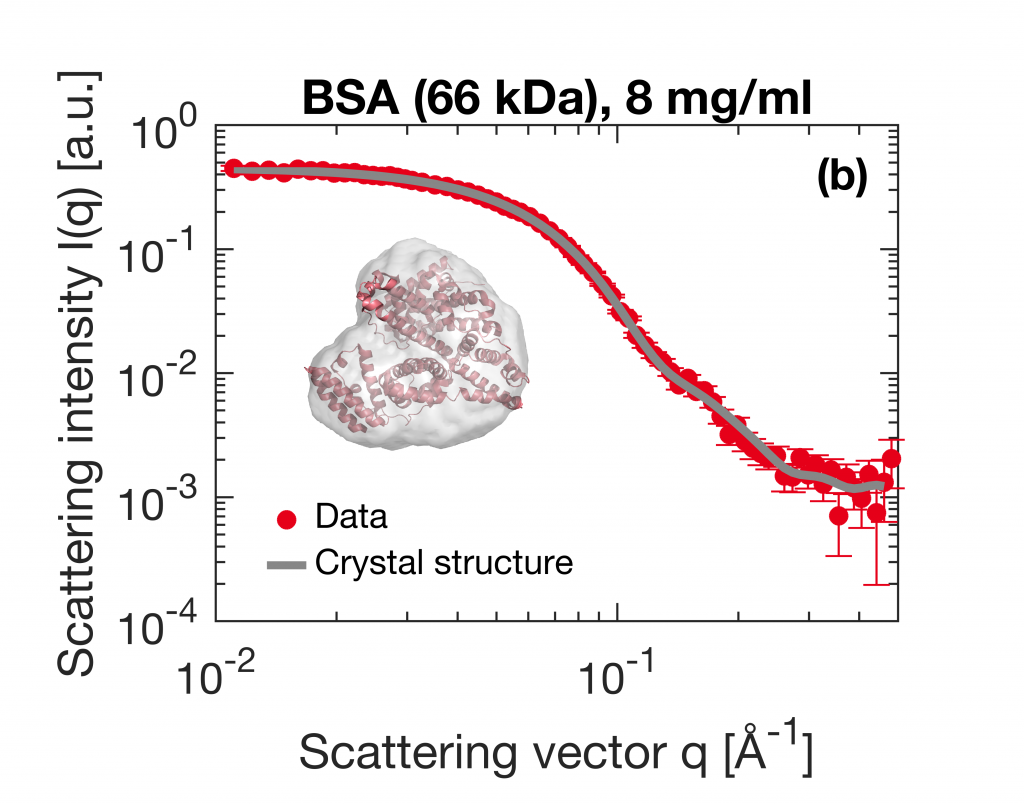
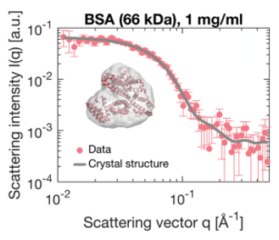
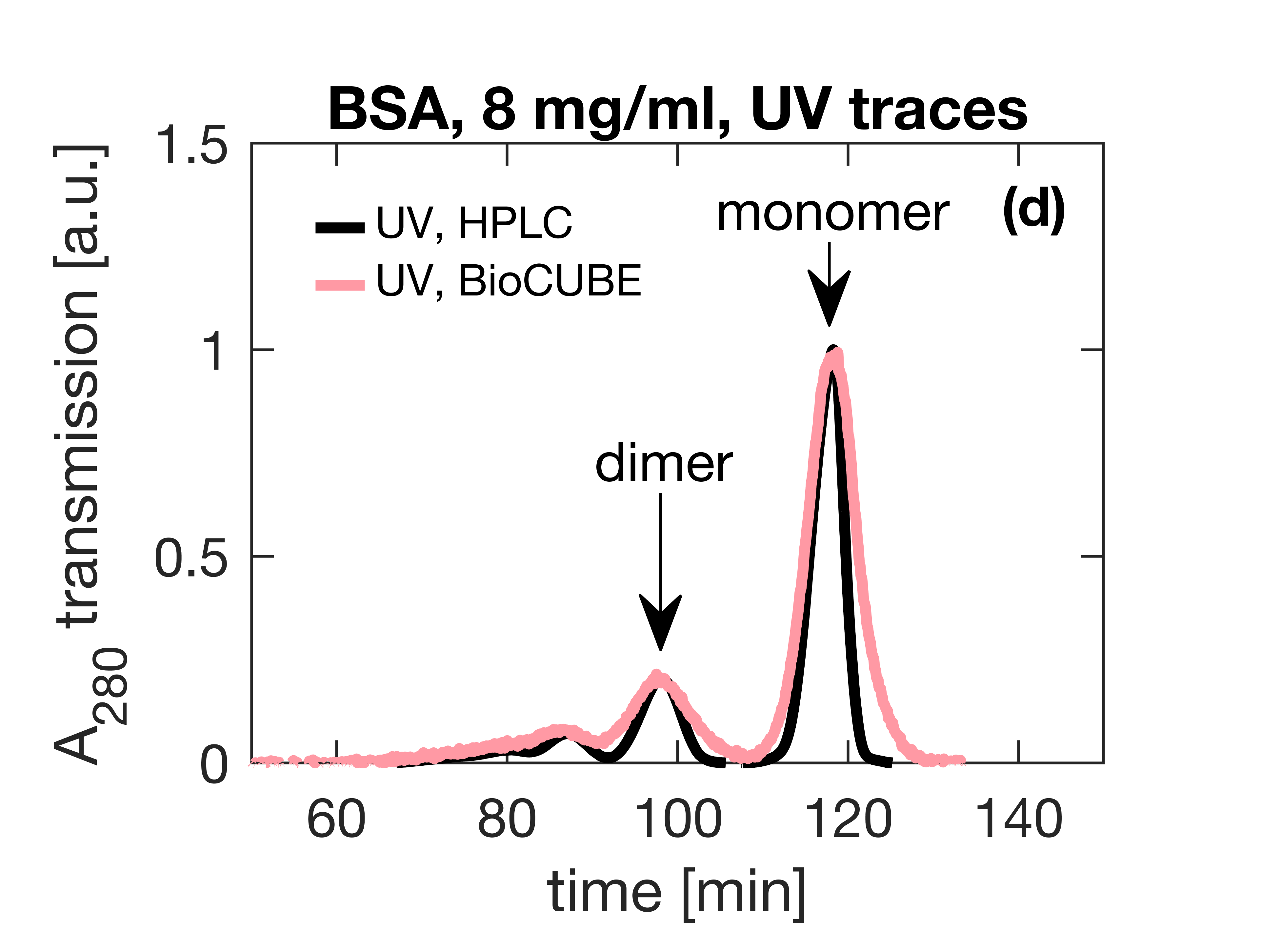{kind=link}
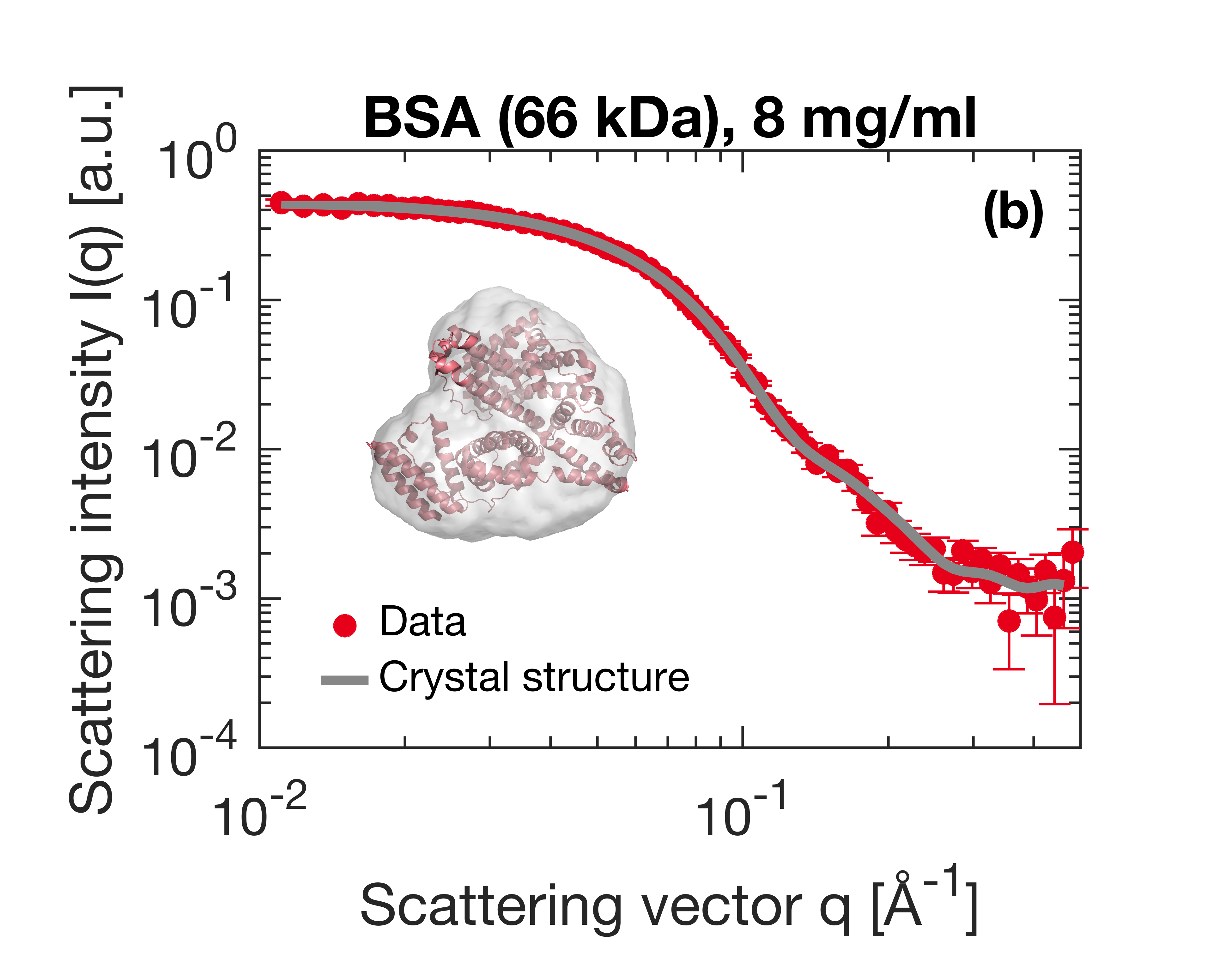{kind=link}
Application note kindly provided by Application Scientist at Xenocs SAS using a BioXolver equipped with Metaljet X-ray source.
Bucciarelli, S. et al. Size-exclusion chromatography small-angle X-ray scattering (SEC-SAXS) of water soluble proteins on a laboratory instrument, J. Appl. Crystallogr. 2018
Using SAXS to gain a deeper understanding of a biomarker for atherosclerosis
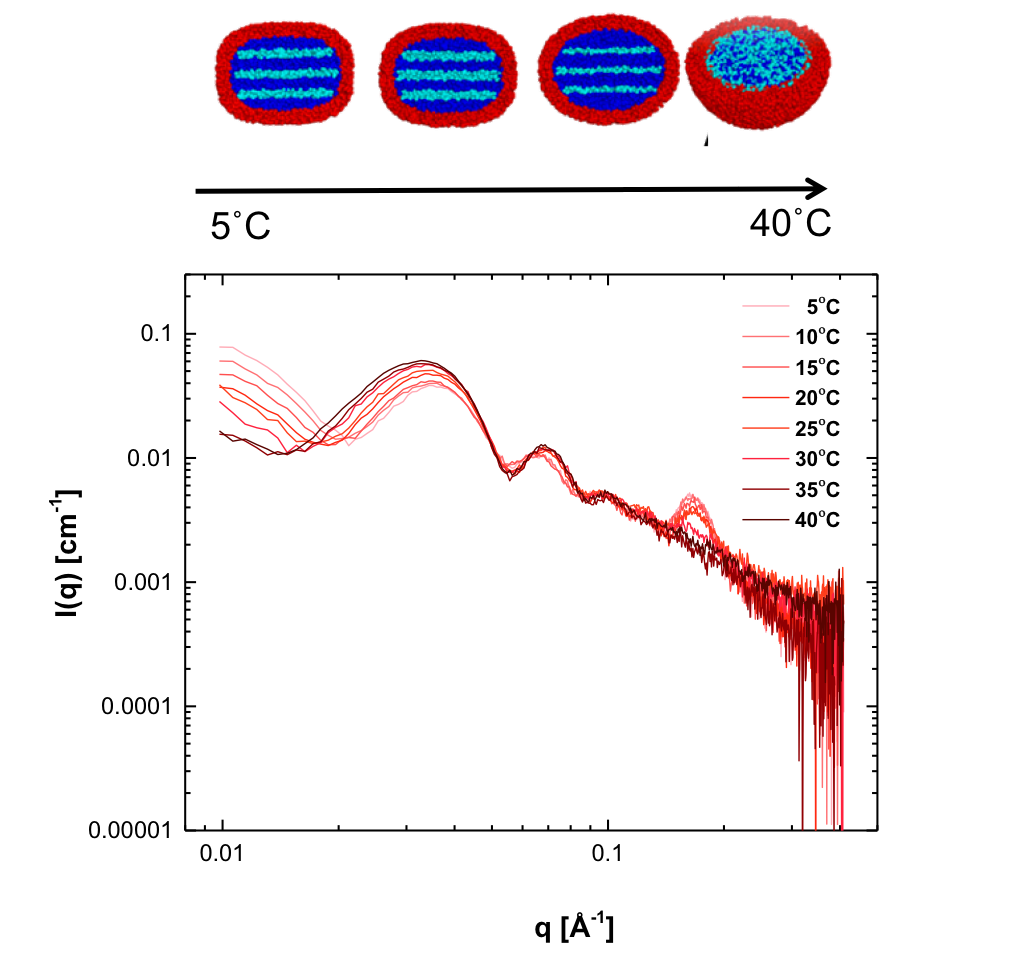
Radially averaged SAXS patterns of LDL, collected at temperatures between 5 and 40˚C.
Low density lipoprotein (LDL), commonly known as cholesterol, is used as a biomarker for atherosclerosis (the narrowing of arteries due to the formations of lesions). Over the past years however, it has become clear that just the amount of LDL found in a patient’s blood is not an accurate predictor of the risk for atherosclerosis. Low density lipoprotein is a mixture of lipids, triglycerides, and sterols of which the structure and composition is different across individuals. Insight into the different LDL components and their structure is crucial for understanding and prevention of atherosclerosis. Complex biomolecular assemblies are some of the most challenging systems to characterize in terms of structure and function by means of biophysical characterization techniques. Crystal structures are difficult to obtain due to the difficulty to crystallize the heterogeneous samples, and Cryo-TEM (cryogenic Transmission Electron Microscopy) requires freezing the species into a fixed conformation. By contrast Small-Angle X-ray Scattering (SAXS) measurements can be done under (near) physiological conditions and offers the possibility of in-situ studies such as under influence of temperature changes, oxidation and even the addition of possible anti-atherosclerotic drugs. Traditionally, this type of measurements could only be done at synchrotron facilities, but the introduction of high brilliance sources means that this can now be done in-house as demonstrated by Maric et al. in a collaboration between Malmö university and Aarhus university. The researchers developed a model describing LDL particles based on temperature dependent SAXS (see picture) and cryo-TEM data. This model successfully describes the size and shape as well as the lamellar internal arrangement of LDL particles and the transition temperature at which the internal structure disappears. Previous studies have shown that the temperature induced transition of the internal structure of the particles is patient dependent and that the ordered arrangement of the core lipids has an effect on the resistance to oxidation, which is known to worsen atherosclerosis. The model was successfully used to describe LDL particles obtained from human blood samples and is expected to enable comparisons between different risk groups in the future.
Fast data collection
As an example of a fast data collection, applications scientists at Bruker AXS recorded data on a crystal of a cyclin-dependent kinase (CDK) using a MetalJet X-ray source mounted on a Bruker D8 VENTURE system. The complete experiment lasted 200 seconds and consisted of 100° of data with the resulting 1.95 Å data allowing for a structure solution by molecular replacement.
- Exposure: 1 second
- Crystal size: 0.1 x 0.08 x 0.05 mm3
- Completeness: 97.5%
- Multiplicity: 3.68
- Rmerge : 6.58%
- Rpim : 3.58%
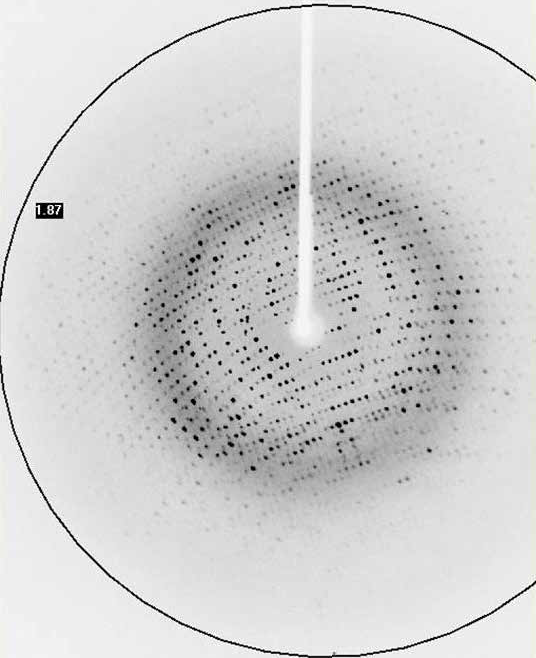
Membrane proteins in-house
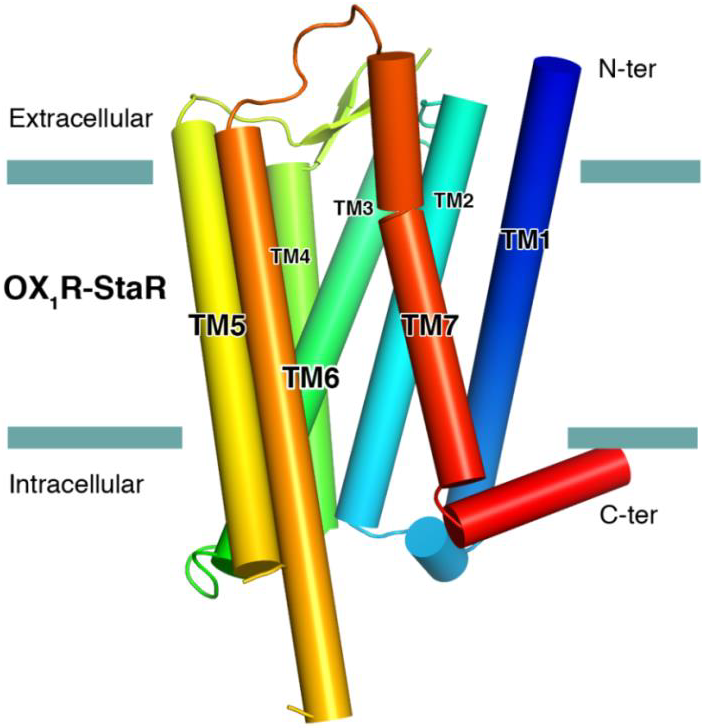
The successful data collection and structure solution of membrane proteins is notoriously difficult and is rarely achieved. Even rarer is the successful determination of a membrane protein structure using an in-house X-ray diffractometer system, rather than a high brilliance synchrotron radiation source. A small crystal of GPCR (Human Orexin receptor Ox1R-StaR®) has been successfully measured at Bruker AXS using an in-house MetalJet X-ray source mounted on a D8 VENTURE diffractometer. Data were collected in a total experiment time of ~2.5 hours to 2.77 Å resolution and the structure was successfully solved by molecular replacement.
- Scan width: 0.1°
- Exposure time: 6 seconds
- Crystal size: 0.08 x 0.08 x 0.05 mm3
- Multiplicity: 3.2
- I/sigma: 8.0
- Rpim : 7.62%
- Rwork/ RFree : 0.244 / 0.272