Protein crystallography
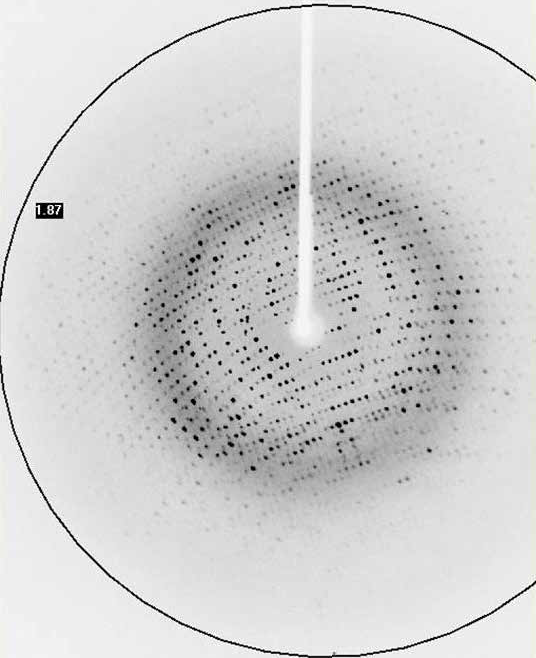
At the same time, the number of atoms present in a protein structure is extremely large and the data (Bragg reflections) to be collected is very closely packed together.
Protein crystallographers rely on the strongest X-ray sources to combat the issues of air sensitivity, small crystals, low diffraction and densely packed reflections. Traditionally, a high brilliance synchrotron has been used to measure full protein data leading to protein structure determination, whilst home laboratory instruments have been used for protein screening to identify the preferred crystals for measurement at the synchrotron.
High brilliance X-ray sources, such as the Excillum MetalJet have made a greater number of protein structures and experiments possible in the home laboratory, thereby accelerating research with ease of access and convenience. Using the high brilliance MetalJet X-ray source makes weak diffraction data stronger, reducing experiment times and potentially reducing sample degradation. The narrow, focused X-ray beam is ideally suited to measuring even the smallest protein crystals, providing compact and well-defined reflections. The higher intensity MetalJet X-rays typically extend the angular resolution limit of the visible protein data collected and provide more precise reflection positions and intensities, leading to higher resolution protein structures.
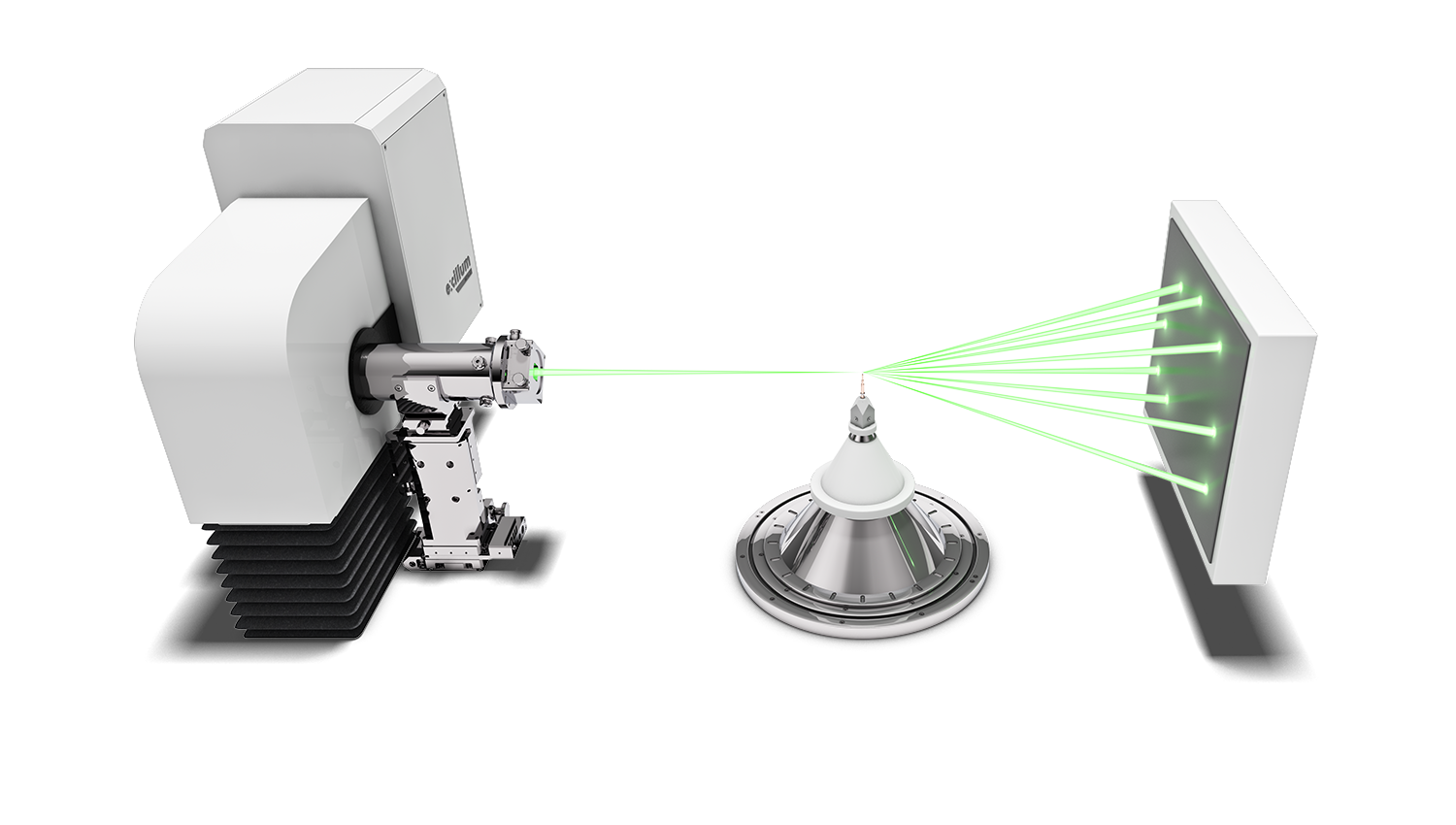
The X-ray source helps a lot, in some cases its use replaces the need for synchrotron. Our main goal with purchasing and using the MetalJet is focused on studying smaller crystals. Working with 30 micron crystals is no longer an obstacle.
Dr. Jan Dohnalek, Institute of Biotechnology, Czech Academy of Sciences, BIOCEV
Application examples for Small molecule crystallography
- Fast data collection
- Membrane proteins in-house
- Ligand-enzyme
- SAD phasing in-house
- In-situ X-ray crystallography
Fast data collection
As an example of a fast data collection, applications scientists at Bruker AXS recorded data on a crystal of a cyclin-dependent kinase (CDK) using a MetalJet X-ray source mounted on a Bruker D8 VENTURE system.
The complete experiment lasted 200 seconds and consisted of 100° of data with the resulting 1.95 Å data allowing for a structure solution by molecular replacement.
- Exposure: 1 second
- Crystal size: 0.1 x 0.08 x 0.05 mm3
- Completeness: 97.5%
- Multiplicity: 3.68
- Rmerge : 6.58%
- Rpim : 3.58%
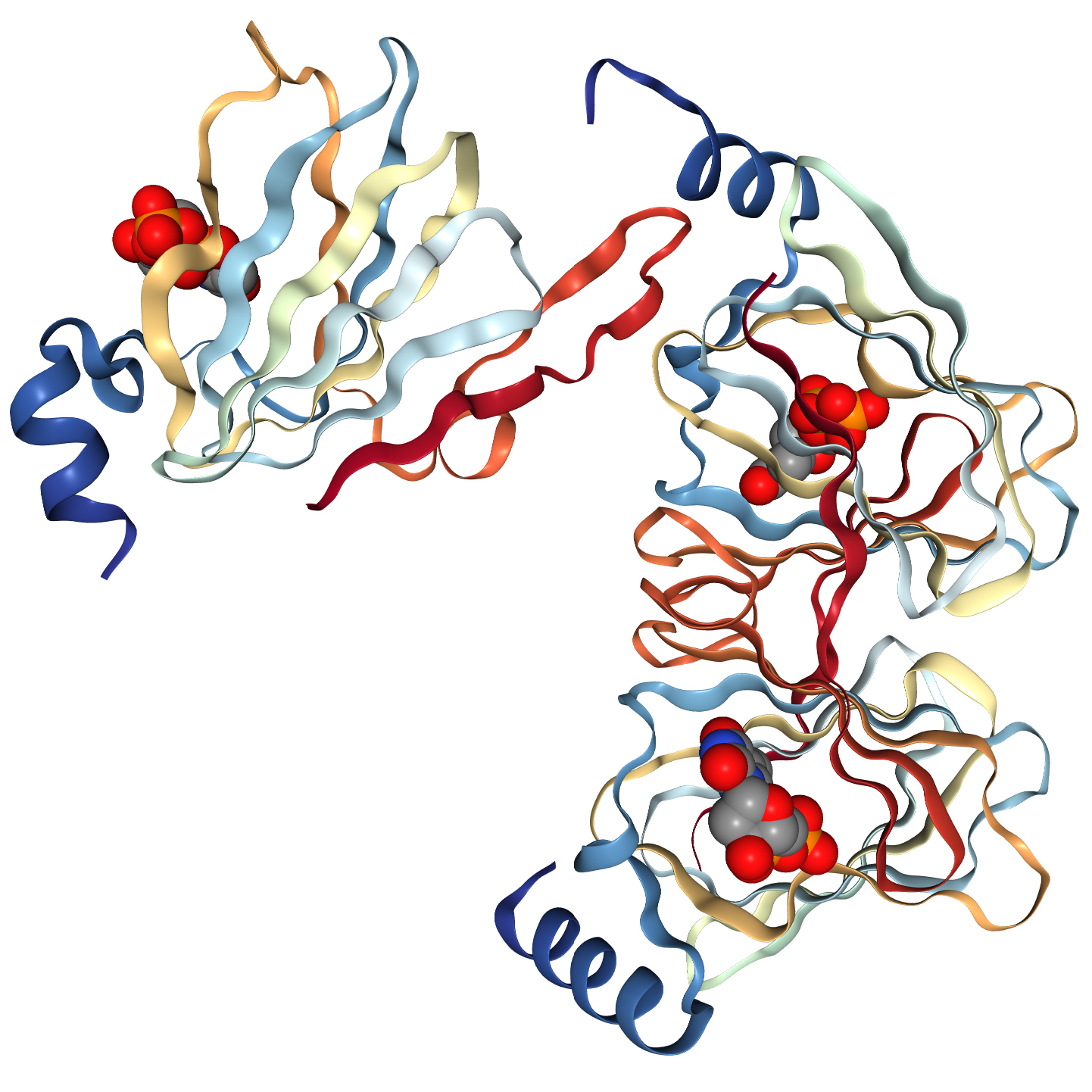
Membrane proteins in-house
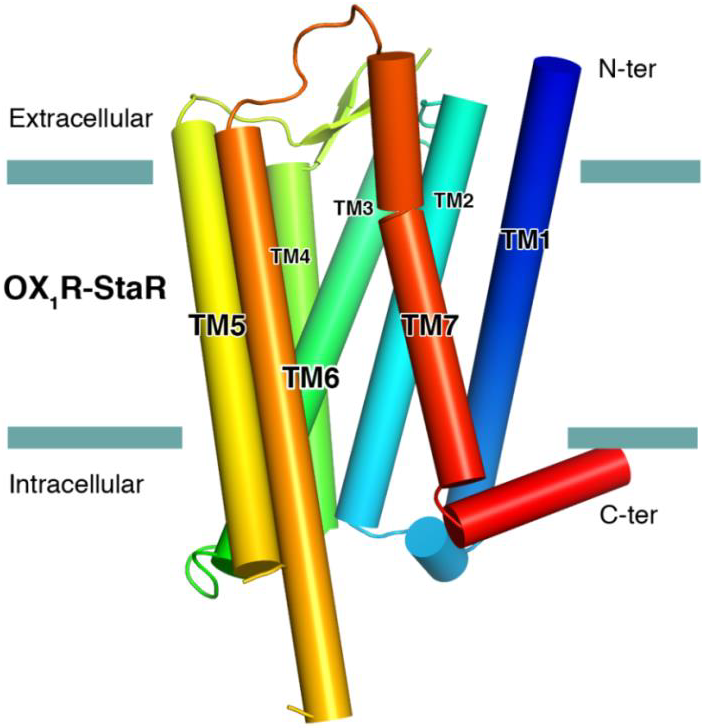
The successful data collection and structure solution of membrane proteins is notoriously difficult and is rarely achieved. Even rarer is the successful determination of a membrane protein structure using an in-house X-ray diffractometer system, rather than a high brilliance synchrotron radiation source.
A small crystal of GPCR (Human Orexin receptor Ox1R-StaR®) has been successfully measured at Bruker AXS using an in-house MetalJet X-ray source mounted on a D8 VENTURE diffractometer.
Data were collected in a total experiment time of ~2.5 hours to 2.77 Å resolution and the structure was successfully solved by molecular replacement.
- Scan width: 0.1°
- Exposure time: 6 seconds
- Crystal size: 0.08 x 0.08 x 0.05 mm3
- Multiplicity: 3.2
- I/sigma: 8.0
- Rpim : 7.62%
- Rwork/ RFree : 0.244 / 0.272
Ligand-enzyme co-crystallisation studies
Researchers at the University of Wisconsin (Department of Biochemistry) and the National Research Council Canada, Human Health Therapeutics, recently solved the structure of WlaRA (TDP-fucose-3,4-ketoisomerase) from Campylobacter jejuni using data collected with a gallium MetalJet D2+ X-ray source.
- Resolution: 2.15 Å
- Completeness: 99.3%
- Rmerge : 7.1%
- Multiplicity: 7.1
SAD phasing in-house
Application scientists at Bruker AXS have determined the crystal structure of Thaumatin obtained from Thaumatococcus danielii by sulphur-SAD phasing methods using data collected in-house on a D8 VENTURE diffraction system with a MetalJet X-ray source.
Using one 70 μm crystal, a complete data set was collected to 1.65 Å in <3 hours. The experimental phases were derived from the anomalous signal of the sulphur atoms and these allowed 95% of the protein backbone to be traced.

In-situ X-ray crystallography
In-situ crystallography is a technique in which protein crystals undergo X-ray diffraction screening and or data collection whilst in a multi-well crystallisation plate and their original growth media/conditions. Typically, X-rays are directed from one side of the multi-well plate, pass through the plate and crystal and out the opposite side of the plate, where the diffraction data are collected on an X-ray sensitive detector.
In-situ X-ray diffraction offers the following benefits:
- Automated, rapid screening and identification of large numbers of potential protein crystals without risk of damage to the crystals
- The identification of protein crystals from non-crystalline objects, salt crystals and other impurity crystals
- The identification of the best protein crystals for further X-ray study
Multi-well crystallisation plates are typically of plastic construction and exhibit low X-ray transparency, high X-ray absorption and significant X-ray background scatter, all of which serve to reduce and/or obscure the X-ray diffraction signal to be studied.
The MetalJet is the ideal choice of X-ray source for in-situ X-ray diffraction in the home laboratory due to the following unique combination of technical features:
- The potential to tune the X-ray beam size through the software, means the X-ray beam may be matched to the size of the crystal and precisely focused on to the crystal of interest, rather than a group of crystals. This also means that a much smaller area of plastic plate is illuminated by the X-ray beam leading to reduced background scatter.
- The higher X-ray brilliance of the MetalJet combined with the lower X-ray absorption of gallium radiation; when compared with copper X-ray sources, means that the X-ray signal obtained from in-situ diffraction is greater for the MetalJet.
